LA LINFOISTIOCITOSI Emofagocitica
HLH
La Linfoistiocitosi Emofagocitica (in inglese Hemophagocytic Lymphohistiocytosis, HLH) è una patologia potenzialmente letale caratterizzata dall’attivazione incontrollata della risposta immunitaria mediata dai linfociti T citotossici, dalle cellule “Natural killer” (NK) e dai macrofagi, causando sintomi e segni infiammatori sistemici. HLH si distingue da altri disturbi infiammatori per la smisurata risposta immunologica caratterizzata dalla iperproduzione di citochine con lesioni a carico di più organi.
Meccanismo molecolare
In circostanze fisiologiche normali, quando le cellule T e NK incontrano un virus o una cellula tumorale, rilasciano granuli citolitici contenenti delle proteine, la perforina e i granzimi che promuovono rispettivamente la formazione di pori (perforina) nella membrana e la distruzione del virus o della cellula malata (i granzimi). Affinché questo processo proceda normalmente, la perforina e i granzimi devono essere proteine strutturalmente normali e opportunamente dirette al bersaglio da eliminare. Infatti i granuli citolitici sono rilasciati nella sinapsi immunologica tra le cellule T e NK e il suo bersaglio, dopodiché il contenuto dei granuli penetra nel bersaglio che è distrutto.
L’interruzione di questo processo di eliminazione del bersaglio a causa di mutazioni genetiche nei geni che codificano per la perforina e granzimi predispone allo sviluppo di HLH primario o familiare. L’incapacità di eliminare virus o cellule tumorali provoca la persistenza e l’amplificazione della risposta immunitaria. Le citochine infiammatorie rilasciate dalle cellule immunitarie provocano alti livelli di attivazione dei macrofagi con conseguente emofagocitosi, danno tissutale, insufficienza d’organo e altre manifestazioni infiammatorie della sindrome.
L’HLH secondaria o acquisita può derivare da uno stimolo maligno, infettivo o autoimmune in assenza di mutazioni genetiche identificabili. Diversi modelli di attivazione e differenziazione dei linfociti T sono stati osservati nei pazienti con HLH secondaria rispetto a quelli con HLH primaria, il che suggerisce possibili differenze nei meccanismi che portano allo sviluppo della patologia in queste forme. IL fattore scatenante nei linfomi che causano HLH è la produzione di citochine infiammatorie che forniscono lo stimolo iniziale e duraturo per l’attivazione dei linfociti T e NK. Il virus Epstein-Barr (EBV) è l’innesco infettivo più comune sia per l’HLH primario che per quello secondario. L’EBV, che normalmente infetta i linfociti B, può causare HLH associato all’EBV attraverso l’infezione dei linfociti T. Questa infezione determina l’attivazione incontrollata e l’attività aberrante del sistema immunitario.
Forme di HLH
Storicamente, i pazienti sono categorizzati come aventi l’HLH “primaria” causata da un difetto ereditario nella funzione citolitica del sistema immunitario (fHLH), o HLH “secondaria” derivante dall’attivazione immunitaria causata da diversi antigeni, incluse le malattie autoimmunitarie da infezioni persistenti o tumori (sHLH).
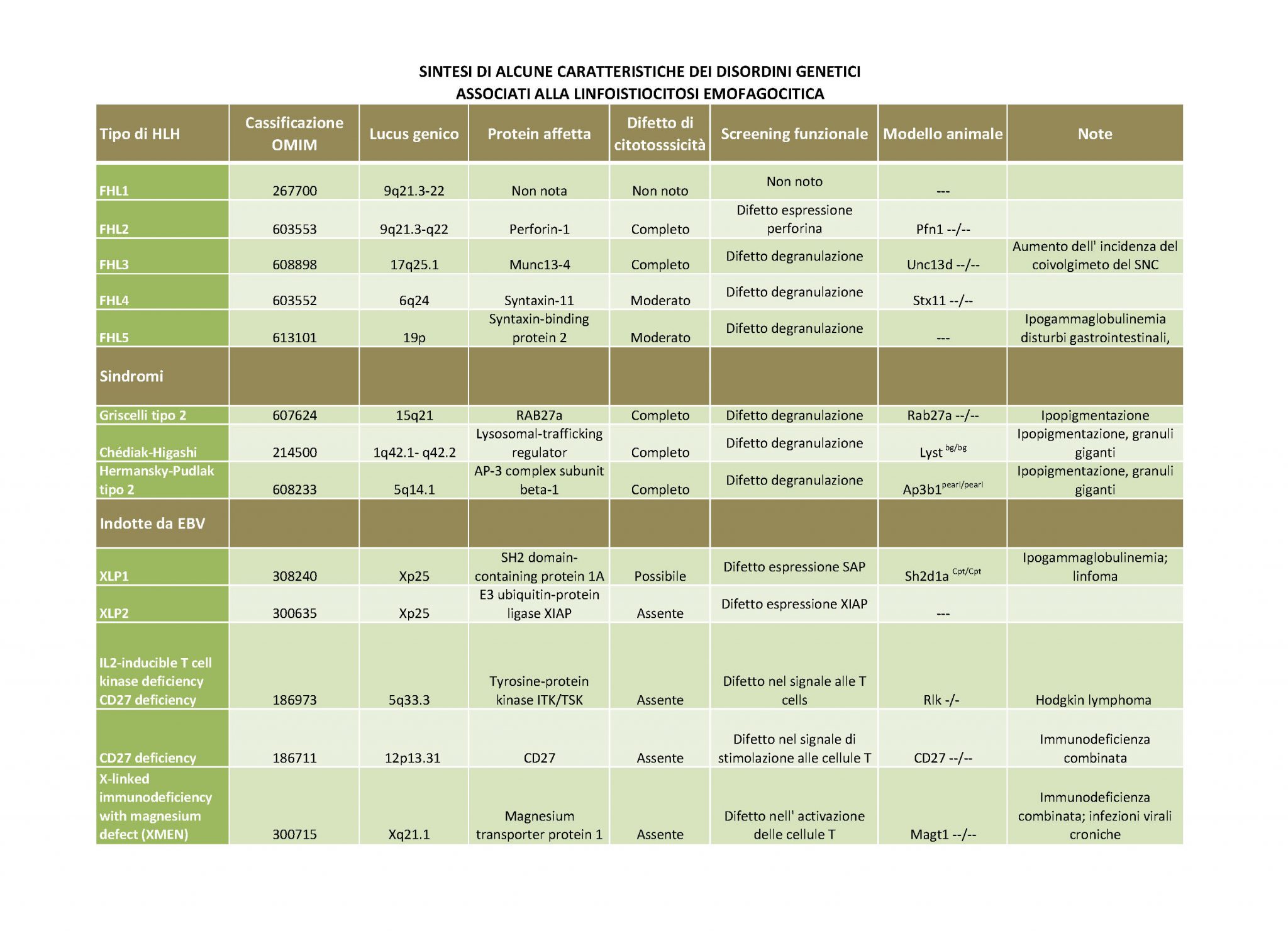
La maggior parte dei bambini con fHLH ha difetti genetici identificabili ed ereditati secondo le leggi mendeliane come lesioni in omozigosi o eterozigosi composte. Questi difetti sono quasi tutte mutazioni che aboliscono l’attività di proteine necessarie per la normale funzione citolitica delle cellule T e NK.
L’HLH secondaria si manifesta negli adulti a causa di fattori scatenanti esterni. Analisi più recenti hanno dimostrato che circa il 15% dei pazienti con sHLH presenta mutazioni nei geni che causano la HLH familiare e che possono servire da fattori predisponenti allo sviluppo di HLH, sebbene ancora in risposta a fattori tipici scatenanti. Questi fattori predisponenti che riducono l’attività di proteine necessarie possono essere un rischio per lo sviluppo di HLH ma di solito non causano la malattia; alcuni di questi fattori occorrono nel 5-10% della popolazione, quindi il loro contributo allo sviluppo di HLH può essere limitato.
A questa visione dicotomica dell’HLH, primaria o secondaria, sta prendendo forma, basata su nuovi dati scientifici, l’idea che la malattia potrebbe essere meglio definita come l’espressione simile di malattie diverse, caratterizzate dall’attivazione immunitaria troppo elevata, da essere tossica, le cui cause sono da imputarsi a meccanismi molecolari diversi, genetici o predisponenti (vedi figura).
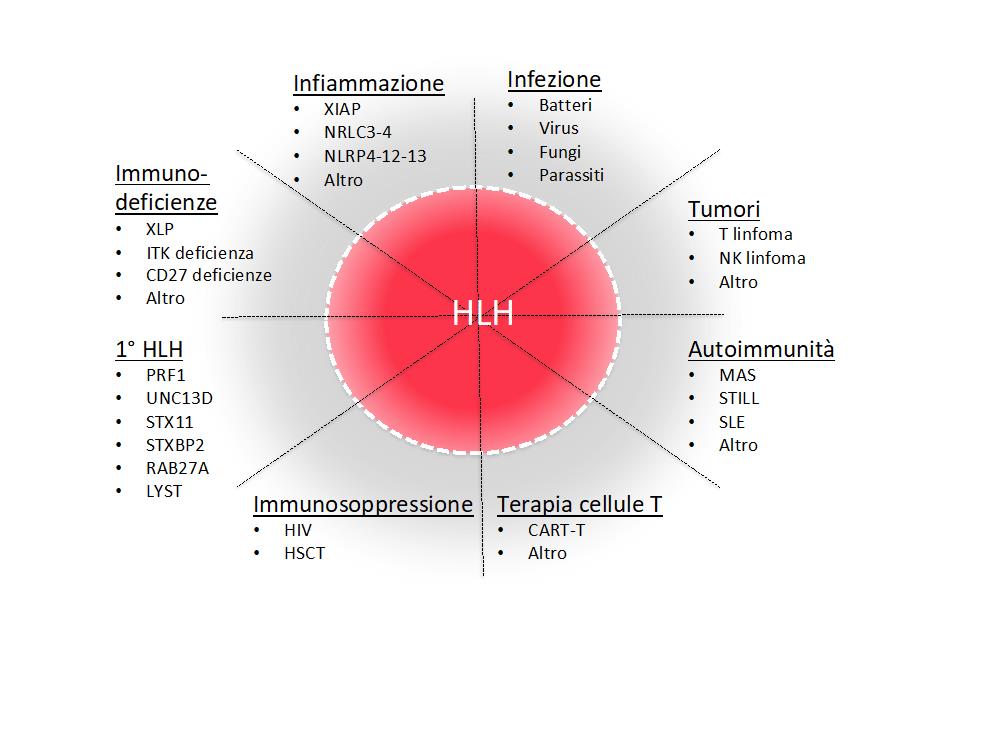
Criteri diagnostici
Clinicamente, l’HLH è una sfida diagnostica perché non esiste alcun sintomo clinico o esame di laboratorio specifico al punto da permettere una diagnosi certa. Le manifestazioni più comuni sono: febbre elevata senza causa apparente, linfoadenopatie, epato-splenomegalia, compromissione della funzionalità epatica, (pan)citopenia, iperferritinemia, ipertrigliceridemia, ipofibrinogenemia, nonché coagulopatia e, in molti casi, manifestazioni neurologiche.
Nel 1994, l’Histiocyte Society americana per poter stabilire dei criteri di inclusione dei pazienti nel primo studio prospettico internazionale, stabilisce 5 principali criteri diagnostici per la diagnosi di HLH: febbre, splenomegalia, bicitopenia, ipertrigliceridemia e/o ipofibrinogenemia ed emofagocitosi. Nel 2004, per il secondo studio internazionale sono introdotti tre criteri aggiuntivi: attività delle cellule NK basse/assenti, iperferritinemia e livelli di recettori dell’interleuchina-2, stabilendo così i criteri diagnostici più comunemente usati e chiamati HLH-2004.
Attualmente la malattia si identifica dopo aver rilevato 5 su 8 dei segni clinici e parametri di laboratorio elencati nella HLH-2004. A questi si aggiunge l’esame morfologico dell’aspirato midollare che consente di confermare la diagnosi e di escludere una leucemia. Inoltre accerta nel 50% dei casi, emofagocitosi cioè la distruzione da parte dei macrofagi di eritrociti, leucociti, piastrine e loro precursori nel midollo e altri tessuti. Infine l’analisi genetica permette di scoprire se alla base della disfunzione immunitaria ci siano eventuali difetti genetici predisponenti o causativi.
HLH e le sindromi Istiocitarie
l’HLH fa parte delle sindromi istiocitarie, un gruppo di malattie causate dalla produzione eccessiva di cellule conosciute come istiociti. Essi includono un’ampia varietà di condizioni che interessano sia i bambini sia gli adulti e sono classificati in tre gruppi in base al tipo d’istiociti coinvolti:
Il primo gruppo è rappresentato da un disordine delle cellule dendritiche, e la malattia più comune in questo gruppo è l’istiocitosi a cellule di Langerhans. Sono incluse anche patologie più rare, come lo xantogranuloma giovanile (JXG) e la malattia di Erdheim Chester.
Il secondo gruppo è rappresentato da un disordine dei macrofagi e include principalmente la HLH e la malattia di Rosai-Dorfman.
Il terzo gruppo è rappresentato dalle istiocitosi maligne e include alcuni tipi di leucemie e tumori.
Terapie per la HLH?
In assenza di trattamento, la Linfoistiocitosi Emofagocitica familiare è rapidamente fatale, con una sopravvivenza mediana di circa 2 mesi dall’esordio. La sopravvivenza a 5 anni è del 10% nei casi trattati con polichemioterapia e del 66% in quelli curati con trapianto di midollo osseo. Per il trattamento della HLH, non vi sono farmaci approvati; esistono raccomandazioni per la gestione della malattia e protocolli di trattamento nazionali e internazionali. E’ importante innanzitutto riconoscere la patologia e improntare tempestivamente non solo il trattamento della stessa ma anche adeguate terapie di supporto al paziente soprattutto nelle fasi iniziali. E’ pertanto indispensabile iniziare una terapia in “mani esperte”, prima che la patologia causi danni irreversibili e/o renda il trattamento meno efficace.
Terapie/trattamenti comunemente usati nella HLH
I pazienti affetti da HLH con un difetto genetico sottostante possono essere solamente trattati quando il sistema immunitario deficitario è sostituito da uno sano che è ciò che accade con il trapianto di cellule staminali ematopoietiche. Alcuni casi di HLH secondaria possono risolversi spontaneamente o dopo trattamento della malattia sottostante. Altri pazienti sono curati con una combinazione di chemioterapia (VP-16, metotrexate), immunoterapia (ATG, ciclosporina) e steroidi. Ogni infezione scatenante deve essere trattata con i famaci antimicrobici appropriati. I pazienti con HLH persistente o ricorrente richiedono, inoltre, un trapianto di cellule ematopoietiche per la guarigione definitiva. Di recente, partendo dall’ipotesi che l’interferon-gamma (IFN-gamma) svolga un ruolo centrale nello sviluppo dell’HLH, è stato avviato un trial clinico con l’obiettivo di valutare l’efficacia e la sicurezza dell’anticorpo anti-IFN nella terapia delle linfoistiocitosi.
Sindrome da attivazione macrofagica
La sindrome da attivazione macrofagica (MAS) è una malattia grave causata dalla produzione eccessiva di una linea di globuli bianchi, chiamati cellule T e macrofagi. La MAS è molto simile alla FHLH e associata ai virus (HLH).
Non è ancora stata determinata l’esatta relazione tra la MAS e la HLH, sebbene alcuni ricercatori credano che la sindrome si sviluppi nel contesto di malattie autoimmunitarie. Il termine è tipicamente utilizzato per la sindrome HLH-simile che può manifestarsi nei pazienti con artrite sistemica ad insorgenza giovanile.
